Universal machine-learning algorithm for predicting adsorption performance of organic molecules based on limited data set: importance of feature description
{kind=link}
Abstract
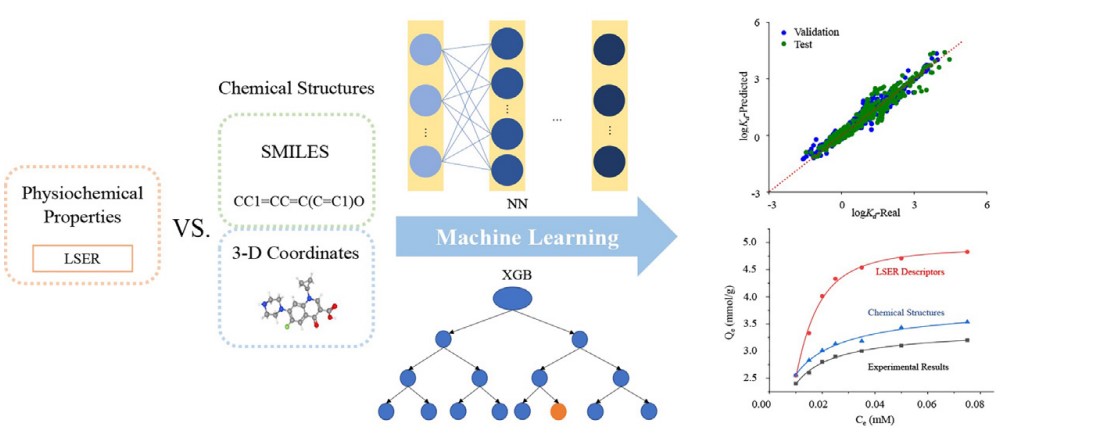
Adsorption of organic molecules from aqueous solution offers a simple and effective method for their removal. Recently, there have been several attempts to apply machine learning (ML) for this problem. To this end, polyparameter linear free energy relationships (pp-LFERs) were employed, and poor prediction results were observed outside model applicability domain of pp-LFERs. In this study, we improved the applicability of ML methods by adopting a chemicalstructure (CS) based approach. We used the prediction of adsorption of organic molecules on carbon-based adsorbents as an example. Our results show that this approach can fully differentiate the structural differences between any organic molecules, while providing significant information that is relevant to their interaction with the adsorbents. We compared two CS feature descriptors: 3D-coordination and simplified molecular-input line-entry system (SMILES). We then built CS-ML models based on neural networks (NN) and extreme gradient boosting (XGB). They all outperformed pp-LFERs based models and are capable to accurately predict adsorption isotherm of isomers with similar physiochemical properties such as chiral molecules, even though they are trained with achiral molecules and racemates. We found for predicting adsorption isotherm, XGB shows better performance than NN, and 3D-coordinations allow effective differentiation between organic molecules.